
Boltz-2, the latest wave in AI models for biology, was announced last week. It’s a leap ahead of its predecessor Boltz-1 which gave Alphafold 3 a run for its money. Where previous models are strong predictors of target structure, binding affinity prediction methods between target and molecule have remained lack-luster. Binding affinity is critical because it tells you how tightly a molecule (drug) binds to its target (protein).
Why it matters in drug discovery:
Binding enables structural change or stabilization, which then leads to protein function (inhibition, activation, etc.). If a molecule doesn’t bind strongly enough, it won’t inhibit, activate, or modulate the protein effectively.
Dosing: Stronger binding usually means lower doses. That’s better for safety, cost, and convenience.
Selectivity: High-affinity binders can be designed to prefer the intended target over other sites, reducing toxicity and unintended effects.
Optimization: Medicinal chemists spend months tweaking compounds to increase affinity. Improvements like this can dramatically speed up drug discovery.
In short: If your compound doesn’t bind well, it usually doesn’t matter what else it can do.
Enter Boltz-2
Boltz-2 slashes this bottleneck by predicting structure and binding affinity, achieving similar results to free energy perturbation (FEP) simulations, which have been the gold standard for binding affinity prediction in drug discovery. While FEP delivers high-precision ΔG estimates rooted in physics, it’s notoriously slow. FEP simulations often require 6–24 hours per run and significant computational resources.
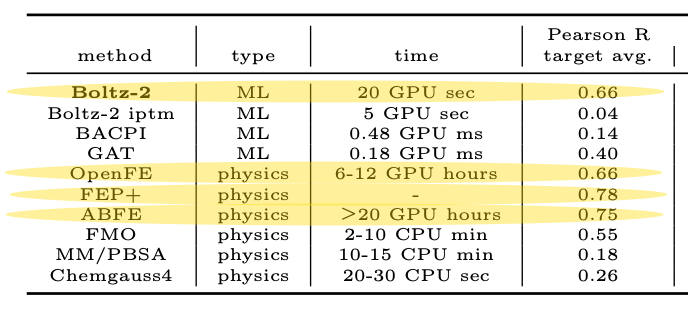
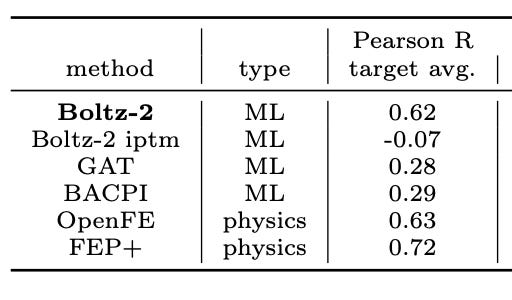
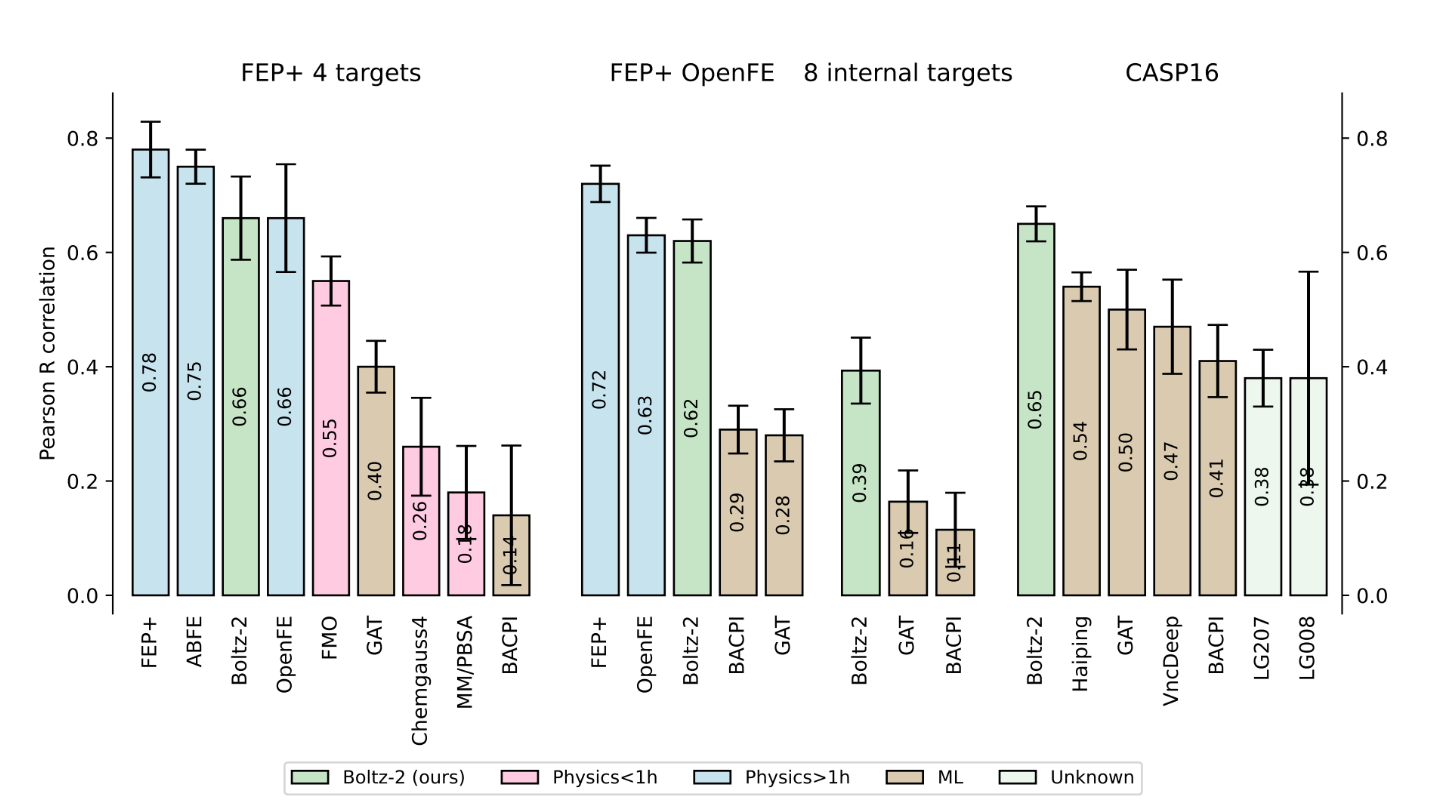
Boltz-2 is the first model to offer a radical alternative. On the FEP+ benchmarks Boltz‑2 hits a Pearson correlation of about ~0.62, matching OpenFE at a fraction of the compute cost, in just seconds (1000x faster). This is what everyone’s excited about.
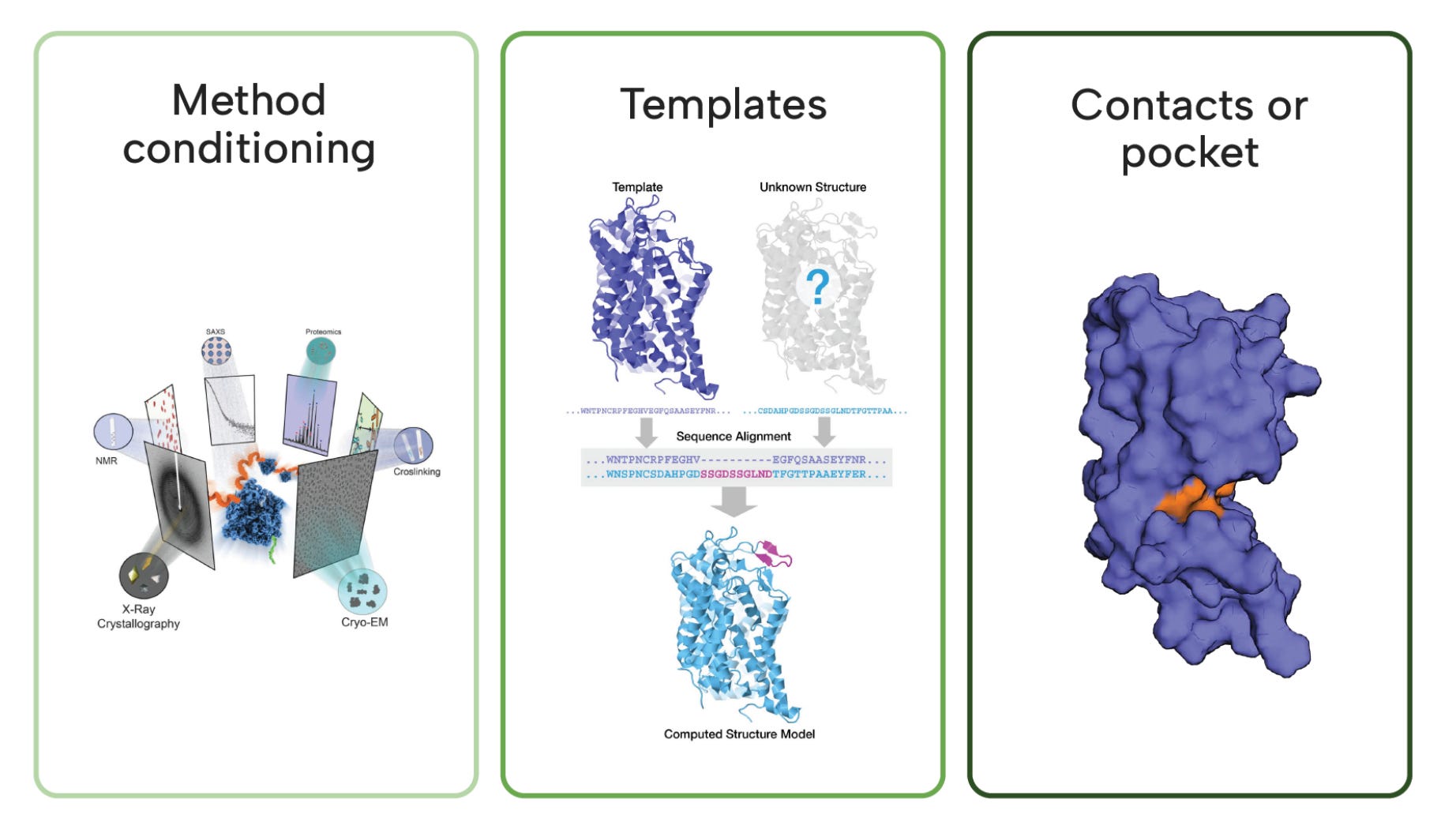
What makes Boltz-2 especially practical is its controllability, making it ready for real-world use. Users can guide predictions at inference time through:
Method conditioning: Users can specify modalities like NMR, X-ray, or molecular dynamics as a conditioning variable to guide structure generation.
Template steering (injecting structural priors): Supports co-folding of multi-chain protein or protein–ligand complexes using templates that embed prior knowledge.
Pocket constraints: You can steer output structures by specifying desired residue-residue or ligand-protein contact maps.
Recursion emphasizes that these added capabilities uphold Boltz-2 as a strong foundation for the next generation of drug discovery pipelines. They’ve already been using Boltz-1 with their proprietary AI models for drug discovery, seeing late discovery programs completed in 18 months instead of the average of 42 months.
What’s under the hood?
Trained on millions of real binding assays (curated from PubChem, PDBBind)
The training set includes ensembles from NMR and molecular dynamics simulations, not just crystal structures. This improves its ability to generalize across flexible systems and binding environments.
Architecture adds an affinity module and supports the steering capability mentioned above.
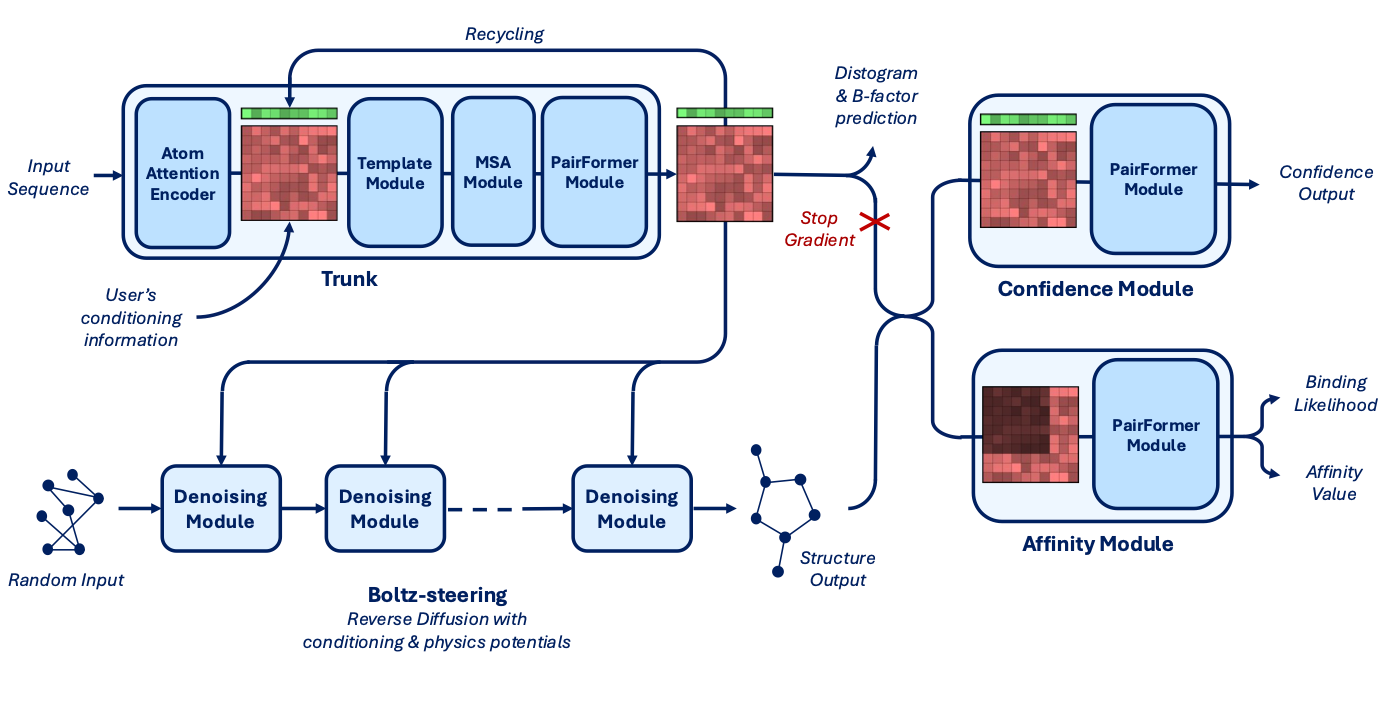
Core Components:
Trunk: Main structural encoder, optimized with
bfloat16and thetrifasttriangle attention kernel to reduce memory and increase token capacity (up to 768 tokens, just like Alphafold 3).Denoising + Steering Module:
Handles masked input recovery and integrates controllability features (as covered above).
Integrated Boltz-steering (from Boltz-1x) at inference time to reduce steric clashes and improve physical plausibility.
Confidence Module: Estimates uncertainty in predicted structures.
Affinity Module: Predicts both binding likelihood and continuous affinity values using a PairFormer model, trained on Ki, Kd, IC50.
Where Boltz‑2 shines and where it doesn’t
Strengths
Speed + Precision: Get docking‑level insight with FEP‑grade accuracy in seconds
Controlled predictions: You can tell it what contacts and pockets to respect
Open‑source + usable: The Boltz-2 model, weights, and training pipeline are available for academic and commercial use. That means teams can fine-tune it for their own modalities, something you can’t (easily) do with AlphaFold 3.
According to Regina Barzilay, pharma groups are already putting serious resources into customizing Boltz-2 for their pipelines.
Key Limitations
Boltz‑2 currently supports binding affinity prediction only for protein–small molecule interactions. It doesn’t yet support protein–protein, protein–peptide, or multi-ligand systems, but it is on the roadmap.
Affinity predictions break down if the model fails to localize the binding pocket or reconstruct the correct protein-ligand conformation. The current version also lacks explicit handling of cofactors like ions, waters, or multimeric binding partners.
Affinity predictions are also limited by the model’s crop size. Important long-range interactions or non-canonical binding pockets may be missed entirely.
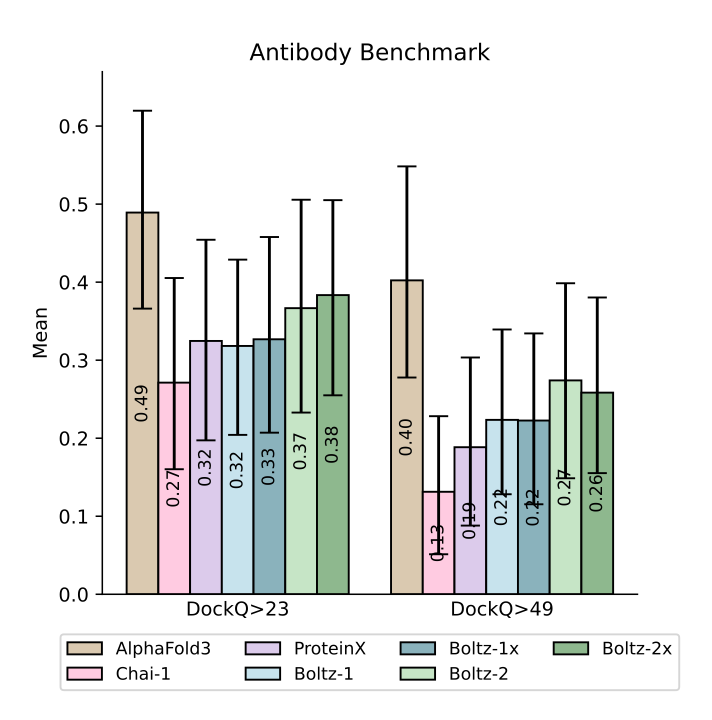
Figure 4 - Boltz-2 paper Worth noting it does not make any ground-breaking progress on the structure prediction side, especially on the antibody benchmark where Alphafold 3 is the clear winner. Although it has made improvements from Boltz-1.
The Boltz team evaluated the model on eight blinded internal assays from Recursion that reflect complex real-world medicinal chemistry projects. Performance on Recursion’s internal assays was mixed: strong on some, weaker on others, highlighting the need for custom input preparation, much like typical FEP methods.
To maximize results, users should:
Define the correct binding region
Include key cofactors or subunits if needed
Steer the model using constraints or known contacts
For Users
These limitations don't undermine Boltz‑2’s breakthrough performance in ligand-centric virtual screening, but they do define its current scope. For tasks involving large structural rearrangements or non-ligand modalities, the model should be supplemented with physics-based approaches or await future extensions.
TL;DR
Boltz‑2 is the first open‑source deep learning model that can both fold structures and predict how well they stick. It’s not a full MD replacement, but for screening and early optimization, Boltz-2 is a viable alternative to FEP simulations, offering FEP-comparable insight at a fraction of the cost. And with built‑in steering, multi‑chain support, and open-sourced code & weights, it’s meant for customizability to real-world workflows.
Run Boltz-2 today + more resources
Run on Google Colab (credit to Aarshit Mittal)
Quick setup, code-commented walkthrough. Good if you want a quick run-through with minimal effort on your end.
You can run Boltz-2 diectly on the BioNeMo website, deploy the NIM in a docker container, or obtain an API key. The NIM has up to 2x acceleration. Great for customizability and integration into your own workflows, production ready.
You can also run Boltz-2 directly on Tamarind Bio. They make it easy to access models with no setup, containers, GPU worries, etc. Similar service to BioNeMo NIMs, perhaps more tailored for those who prefer a simpler, no-code environment.
More resources:



